一、小儿遗传性共济失调的发病原因有哪些
1、发病原因
1863年描述的Friedreich共济失调(FRDA)是一种最常见的早发型常染色体隐性共济失调,最基本的临床表现为青少年起病(青春期到25岁之间),进行性步态和肢体共济失调,腱反射缺失,跖反射伸性反应,其他常见特征为构音障碍,皮质脊髓束性笨拙,腿部本体感觉功能缺失,脊柱侧凸和心脏病,25岁以前起病的腿部腱反射缺失的Friedreich共济失调,要与迟发的Friedreich共济失调及其他诸如伴有腱反射缺失的早发性小脑性共济失调等“Friedreich共济失调样”综合征区分开来,随着Friedreich共济失调基因被克隆,已经明确某些“Friedreich共济失调样”综合征,也是Friedreich共济失调基因突变的结果。
临床上应用较多的分类如下:
1)脊髓型包括:
1.弗里德里希共济失调(Friedreichsataxia)。
2.遗传性痉挛性截瘫。
3.后柱性共济失调等。
2)脊髓小脑型包括:
1.遗传性痉挛性共济失调。
2.无β脂蛋白血症。
3.共济失调毛细血管扩张症(Louis-Bar综合征);④脊髓脑桥变性等。
3)小脑型包括:
1.橄榄桥小脑萎缩。
2.小脑橄榄萎缩。
3.肌阵挛性小脑协调障碍(Ramsay-Hunt综合征)。
4.Machedo-Joseph病(又称Azorean病)。
5.遗传性共济失调-白内障-侏儒-智力缺陷综合征。
6.Hartnup病等。
由于该分类未包括病因及发病机制,造成与其他分类方法的重叠,例如无β脂蛋白血症及Hartnup病属先天代谢异常,共济失调毛细血管扩张症则又属神经皮肤综合征的范畴。
2、发病机制
1)弗里德里希共济失调(Friedreichsataxia)弗里德里希共济失调首先由Friedreich于1863年报告,是遗传性共济失调中研究较深入的类型,多数病例尤其是典型病例属常染色体隐性遗传,少数病例似属显性遗传,或为散发,本病的病因尚不清楚,亦未发现特异性的生化异常,病变主要累及脊髓后索和侧索中的脊髓小脑后束和锥体束,脊髓小脑前束病变较轻,病理改变主要为神经纤维脱髓鞘及轴索破裂,Clark柱细胞消失,胶质增生,后根也有类似病变,小脑损害相对较轻微或为正常,多数病例伴有心肌纤维的弥漫性变性和结缔组织增生。
2)遗传学Friedreich共济失调的基因(X25)定位于第9号染色体的9q,编码一个高度保守的蛋白,frataxin,95%以上的Friedreich共济失调病人是X25基因第一个内含子的GAA三联子的重复扩增的纯合子,少数Friedreich共济失调病人是一个GAA内含子扩增和截断(truncating)或无义(missense)突变的杂合子,一些但不是全部病人为不典型和轻度病变的复合杂合子。
3)共济失调毛细血管扩张症共济失调毛细血管扩张症,又名Louis-Bar综合征是累及神经,血管,皮肤,内分泌,网状内皮系统等的原发性免疫缺陷病,也是一种染色体不稳定综合征,主要病理改变是弥漫性小脑皮质萎缩,脊髓薄束和脊髓小脑束脱髓鞘,胸腺明显缩小或缺失。
4)橄榄桥小脑萎缩橄榄桥小脑萎缩呈常染色体显性遗传或为散发,病变主要累及橄榄核,脑桥基底核和小脑半球,脊髓后索和脊髓小脑束也可受累,受累部位细胞明显减少,髓鞘脱失。
二、小儿遗传性共济失调有哪些典型症状
1、弗里德里希共济失调
1)本病多在5~18岁发病,少数可迟至30岁发病,隐性遗传者起病较显性遗传者早,同胞的发病年龄相近,男女大致相等,隐匿性起病,病程呈缓慢进行性发展,早期表现为步态不稳,步态蹒跚,站立时身体摇晃,宽基底步态,闭目难立征阳性,肌张力低下,腱反射消失,随病情进展出现双手笨拙,意向性震颤,构音障碍,说话缓慢而含糊,后期出现肢体深感觉消失,因锥体束损害明显而出现病理反射,多数有眼震,少数有视神经萎缩,晚期可出现轻度智力低下。
2)骨骼畸形是本病又一特征,弓形足,脊柱后凸或侧凸尤为常见。
3)胸部X线片常提示心脏扩大,心电图及超声心动图提示心肌病,可发生心力衰竭和心律失常,20%病人发生糖尿病而需行胰岛素治疗,40%~50%病人糖耐量异常,糖尿病易发生于20~30岁病人,其神经系统并发症反过来可加重病人本身的症状。
4)部分病人可伴有白内障,蓝巩膜等,神经传导速度测定显示感觉动作电位明显降低或消失,运动神经传导速度仅轻度减慢,与轴索性神经病的病变特点一致。
2、共济失调毛细血管扩张症
1)男女大致相等或女略多于男,婴幼儿期起病,首发症状是小脑性共济失调,意向性震颤,构音障碍,眼震及步态不稳,随病情进展可出现锥体外系症状及脊髓症状,表现为肌张力异常,手足徐动,深感觉缺失,病理反射阳性等,还可伴发脊柱前凸或侧突,智力逐渐减退,腱反射减弱或消失。
2)毛细血管扩张常在4~6岁出现,最先见于球结膜,以后出现于眼睑,颊部,耳郭,颈部锁骨上部,上肢的屈侧等处皮肤,皮肤及毛发呈早老性改变,女性患者卵巢不发育,病儿常有反复的呼吸道感染,胸腺不发育,约半数并发恶性肿瘤。
3、橄榄桥小脑萎缩
男女比例约为2∶1,发病年龄2个月至60岁,多于30岁左右起病,发病隐匿,病初常觉下肢易倦,步态不稳,双手动作渐不灵活,精细动作难以完成,伴明显的言语障碍,意向性震颤及辨距不良,部分病人出现吞咽困难,继之渐出现帕金森综合征,肌张力由减低变为僵直,除头,肢体及躯干震颤之外,可见腭帆提肌的重复收缩(软腭震颤),间有舌及面部肌束震颤,部分病人有眼肌麻痹,视神经萎缩,眼球震颤,色素性视网膜炎,晚期可有锥体束损害表现,尿失禁及视力障碍,由于快速扫视运动障碍引起慢眼球运动,呈凝视状,晚期可使眼球几乎固定。
三、小儿遗传性共济失调需要做哪些化验检查
1、实验室检查
1)血象检查:
感染时外周血白细胞计数和中性粒细胞分类显著增高。
2)血液检查:
1.免疫球蛋白异常:40%~80%患儿血清和分泌型IgA和IgG缺乏或减少,IgM增高。
2.甲胎蛋白升高:对离子辐射的敏感性异常及α-甲胎蛋白明显升高。
3.细胞遗传学异常:染色体检查可见同源14号染色体易位〔t(14q+;14q-)〕。
2、辅助检查
1)包括特征性的心电图改变和向心室性肥厚或不常见的不对称性间隔肥厚的心动超声证据。周围神经传导速度正常及感觉神经动作电位缺失或显著下降是Friedreich共济失调与Charcot-Marie-Tooth病的区别要点。其他常见的异常为视觉诱发电位的波幅下降,锁骨上(体感诱发电位)降低,或缺如以及感觉皮质的迟发扩散性电位。
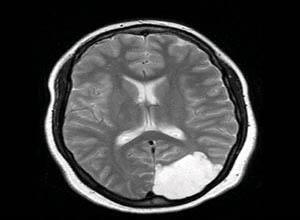
2)脑CT扫描及MRI检查可见小脑及脑干萎缩。MRI常可显示颈髓萎缩。
四、小儿遗传性共济失调病人的护理
1、患者可以适量的吃鸡蛋。鸡蛋中所含的蛋白质是天然食物中最优良的蛋白质之一,而蛋黄除富含卵磷脂外,还含有丰富的钙、磷、铁以及维生素A、D、B等。
2、菠萝、柠檬、香蕉菠萝中富含维生素C和重要的微量元素锰,对提高人的记忆力有帮助;柠檬可提高人的认知能力;香蕉可向大脑提供重要的物质酪氨酸,可使人精力充沛、注意力集中,并能提高人的创造能力。
3、全麦制品和糙米。糙米中含有多种维生素(特别是维生素B1),对于提高患者的认知能力至关重要。
4、患者可以多吃鱼类。鱼肉脂肪中含有ω-3脂肪酸,有助于健脑。吃鱼还有助于加强神经细胞的活动,从而提高学习和记忆能力。
5、核桃和芝麻也是患者应该多吃。核桃和芝麻核可为大脑提供充足的亚油酸、亚麻酸等不饱和脂肪酸,可排除血管中的杂质,提高患者的大脑功能